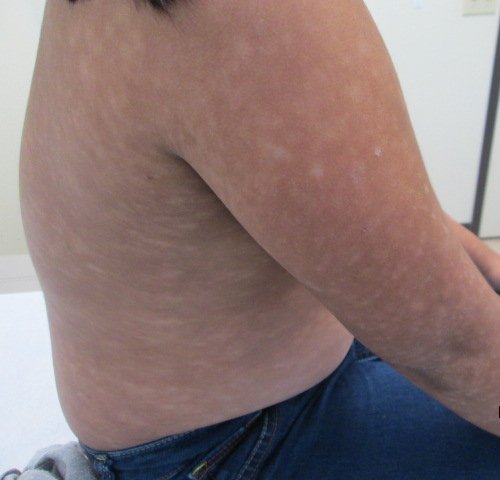
CORRECT DIAGNOSIS:
Hypopigmented mycosis fungoides
DISCUSSION:
Mycosis fungoides (MF) is the most common primary cutaneous T cell lymphoma that usually presents in the stages of patches, plaques, tumors or erythrodermca [1-4]. There are several distinct variants of MF that have their own clinical behavior and outcome. They include pustular, bullous, purpuric, follicular, icthyosiform, veurrucous, unilesional, invisible, granulomatous, hyperkeratotic, and hypopigmented mycosis fungoides (HMF) [2, 5].
HMF is an indolent cutaneous T cell lymphoma that represents a rare clinical variant of the early patch stage of MF [1, 3, 6, 7]. It is most commonly seen in dark skinned and Asian individuals, it has infrequently been reported in Caucasians as well [1-8]. The average age of onset is usually seen between the first and third decades as compared to classic MF, which manifests around the sixth decade [1, 2, 6, 7, 9].
Patients will most commonly present with multiple, ill-defined, hypopigmented patches of varying sizes on the trunk, back and proximal extremities [2, 4-6, 8]. Cutaneous sensation will be intact with a variable degree of scaling, atrophy, infiltration, and pruritus [2, 4-6, 8].
The differential diagnosis for HMF includes all possible etiologies for hypopigmented patches on the skin, such as vitiligo, leprosy, progressive macular hypomelanosis, sarcoid, pitryiasis lichenoides chronica, tinea versicolor, pitryiasis alba, morphea, post inflammatory hypopigmentation, hypomelanosis of ito, syphilis, among many others [1-6, 8].
Histopathological examination usually shows an intense disproportionate epidermotropism of large haloed lymphocytes with convoluted nuclei individually or in small clusters (pautrier microabscesses), basilar tagging of lymphocytes, lymphocytic infiltration in the dermis with mild atypia, papillary dermal fibrosis and wiry collagen [1-9]. Occasionally pigment incontinence, melanophages, and folliculotropsim may also be seen [5, 6, 9]. HMF lesions characteristically demonstrate CD8+ T cells with a low CD4+: CD8+ ratio, unlike classic MF that presents with a CD4+ predominant infiltrate [1-9].
Proposed mechanisms for the cutaneous hypopigmentation include the cytotoxic effect of CD8+ T cells on CD117 causing dysfunction and/or loss of melanocytes, a defect in the transfer of melansomes from melanocytes to keratinocytes, and a nonspecific result of inflammation [2, 4, 8, 9].
The presence of a T-cell receptor gene arrangement is seen in a large percentage of patients with HMF, however the absence of clonality does not rule out the disease [1, 6-9]. Initial evaluation may include CBC with differential, quantification of T and B cell lymphocytes using flow cytometery, physical exam of the peripheral lymph nodes, and diagnostic imaging if suspecting any systemic involvement [2, 7].
Psoralen + UVA (PUVA) or narrow band UVB with/without topical steroids is the mainstay of treatment [1-10]. Topical nitrogen mustard, topical carmustine, electron beam therapy, bexarotene, methotrexate, and pralatrexate have also showed effectiveness [2, 4, 5, 7, 8, 10]. Most patients require repetitive cycles of phototherapy or maintenance therapy due the high recurrence rates associated with HMF [1, 2].
HMF has an indolent course with an overall survival of 98% at 20 years, a significantly better prognosis than classic MF [3, 5]. However there have been cases of progression to tumor, systemic involvement, and even death [3, 4, 9]. It has been postulated that the predominance of CD8+ T cells prevents the evolution of HMF lesions to MF plaques and tumors by participating in the protective Th1 response [2, 3]. Hypopigmentation may be considered as a marker of good prognosis.
The diagnosis is usually rendered by clinicopathological correlation supplemented by immunohistochemistry and ancillary studies such as TCR-gene rearrangement, blood tests, and diagnostic imaging. Occasionally you may need several skin biopsies to confirm the disease.
Due to the increasing frequency of HMF it is important to be aware of this entity and perform a skin biopsy especially in patients with persisting or progressing hypopigmentation despite therapy. HMF is a malignant neoplastic disease with a lethal potential and should be treated as such with proper work up and mandatory follow up.
TREATMENT:
Once the diagnosis of hypopigmented mycosis fungoides was made, patient was referred to oncology for close follow-up and started on a regimen NB-UVB therapy.
REFERENCES:
1. Castano E, Glick S, Wolgast L, Naeem R, Sunkara J, Elston D, Jacobson M: Hypopigmented mycosis fungoides in childhood and adolescence: a long-term retrospective study. J Cutan Pathol 2013, 40(11):924-934.
2. Furlan FC, Sanches JA: Hypopigmented mycosis fungoides: a review of its clinical features and pathophysiology. An Bras Dermatol 2013, 88(6):954-960.
3. Gameiro A, Gouveia M, Tellechea O, Moreno A: Childhood hypopigmented mycosis fungoides: a commonly delayed diagnosis. BMJ Case Rep 2014, 2014.
4. Stone ML, Styles AR, Cockerell CJ, Pandya AG: Hypopigmented mycosis fungoides: a report of 7 cases and review of the literature. Cutis 2001, 67(2):133-138.
5. Hassab-El-Naby HM, El-Khalawany MA: Hypopigmented mycosis fungoides in Egyptian patients. J Cutan Pathol 2013, 40(4):397-404.
6. Koorse S, Tirumalae R, Yeliur IK, Jayaseelan E: Clinicopathologic profile of hypopigmented mycosis fungoides in India. Am J Dermatopathol 2012, 34(2):161-164.
7. Tolkachjov SN, Comfere NI: Hypopigmented mycosis fungoides: a clinical mimicker of vitiligo. J Drugs Dermatol 2015, 14(2):193-194.
8. Khopkar U, Doshi BR, Dongre AM, Gujral S: A study of clinicopathologic profile of 15 cases of hypopigmented mycosis fungoides. Indian J Dermatol Venereol Leprol 2011, 77(2):167-173.
9. El-Shabrawi-Caelen L, Cerroni L, Medeiros LJ, McCalmont TH: Hypopigmented mycosis fungoides: frequent expression of a CD8+ T-cell phenotype. Am J Surg Pathol 2002, 26(4):450-457.
10. Galper SL, Smith BD, Wilson LD: Diagnosis and management of mycosis fungoides. Oncology (Williston Park) 2010, 24(6):491-501.